Target enrichment with hybridization capture
Hybridization capture targets regions of the genome specific to researchers’ objectives using 5’ biotinylated probes. After isolating probes annealed to complementary fragments with streptavidin-coated beads, the library fragments are sequenced. The resulting data focus on the targeted regions, providing more read depth for variant identification.
xGen™ NGS—made for hybridization capture.
Overview
- Customizable—select between a small panel or a large panel depending on your research needs
- Probe design—tile probes across the entire region of interest or target the ends of the region
- Expandable—increase your targeted regions of interest by adding probes to existing hybridization capture panels
- Focused data—increase likelihood of rare variant identification
What is hybridization capture?
Hybridization capture, often called target enrichment, lets researchers reliably target large numbers of genes specific to their research objectives by using complementary 5’ biotinylated probes that anneal and separate the specific genomic DNA library fragments from non-specific library fragments.
This method provides data for the discovery of novel variants because hybridization capture targets a higher amount of total gene content. The subsequent profiling of the variant types is comprehensive and provides the basis for thorough characterization of the newly identified variant.
Hybridization capture applications:
- Exome sequencing
- Genotyping SNPs or indels
- Pan-cancer or inherited disease research
- Identification of rare variants
Hybridization capture works well for genotyping and rare variant identification. Since capture with hybridization probes does not require PCR primer design, it is less likely to miss mutations and performs better with respect to sequence complexity [1]. Hybridization capture’s capacity for mutation discovery makes it particularly suited to cancer research. Both its sequence complexity and scalability make it an excellent choice for exome sequencing. Depending on your sample type or experimental goals, you can use UMIs (unique molecular identifiers), sometimes called ‘molecular barcodes.’ These complex indexes can be added to sequencing libraries before the PCR amplification steps to enable the correct bioinformatic identification of PCR duplicates. UMI sequence information along with alignment coordinates permit grouping of sequencing data into read families that represent individual sample DNA or RNA fragments.
For a deeper look into how the method is performed, see the IDT Targeted sequencing guide.
Hybridization capture workflow
Extraction
Sequencing & analysis
Method data
Hybridization capture of FFPE DNA and cfDNA samples
We extracted DNA from matched trios of biobank sourced material from three individuals which included adjacent fresh-frozen normal tissue, matched formalin-fixed paraffin-embedded- (FFPE)-tumor tissue, and plasma samples (Figure 1). The AnaPrep FFPE DNA Extraction Kit (BioChain) and the cfPure® V2 Cell-Free DNA Extraction Kit (BioChain) were used to perform DNA extraction. Depending on the sample, one of the following methods was used to assess its quality:
- Fluorometric quantification—Qubit™ dsDNA BR Assay Kit (Thermo Fisher Scientific)
- Capillary electrophoresis—Bioanalyzer® HS DNA chip (Agilent)
- qPCR—KAPA® hgDNA Quantification and QC Kit (Roche)
Sequencing libraries were generated with 100 ng of DNA extracted from the FFPE samples and adjacent fresh-frozen normal samples. Despite low-input sample quality, the xGen cfDNA & FFPE DNA Library Prep Kit generated high-yield libraries.
The resultant libraries were captured in single-plex with a custom xGen Pan-Cancer hybridization panel and sequenced. Picard (Broad Institute) was used to evaluate library preparation and hybrid capture results including hybrid-selection (HS) library size (Figure 2), duplicate rate, and coverage after standard start-stop deduplication. Our experimental goal was to identify as many variants as possible.
High-yield libraries were generated with 25 ng of cfDNA isolated from the matched plasma from each of the three individuals and captured with subject-matched custom xGen Custom Hyb Panels. Incorporation of unique dual indexes (UDIs) increased resolution and prevented sample misassignment. Despite the small size of these panels, we obtained high on-target rates and a sequence depth sufficient to reach duplication rates of >80%, which is recommended for collapsed read analysis to enable error correction
(Figure 3A). Collapsing reads uses unique molecular identifiers (UMIs) to remove sample prep, library prep, and sequencing errors, which leads to reliable variant calling of ultra-low frequency variants.
Mapped reads were used to generate
collapsed single read families, as outlined in the xGen cfDNA and FFPE DNA Library Prep Kit Analysis Guidelines. The combination of the xGen cfDNA & FFPE DNA Library Prep Kit with xGen Custom Hyb Panels capture resulted in high complexity (Figure 3A) and coverage for cfDNA (Figure 3B).
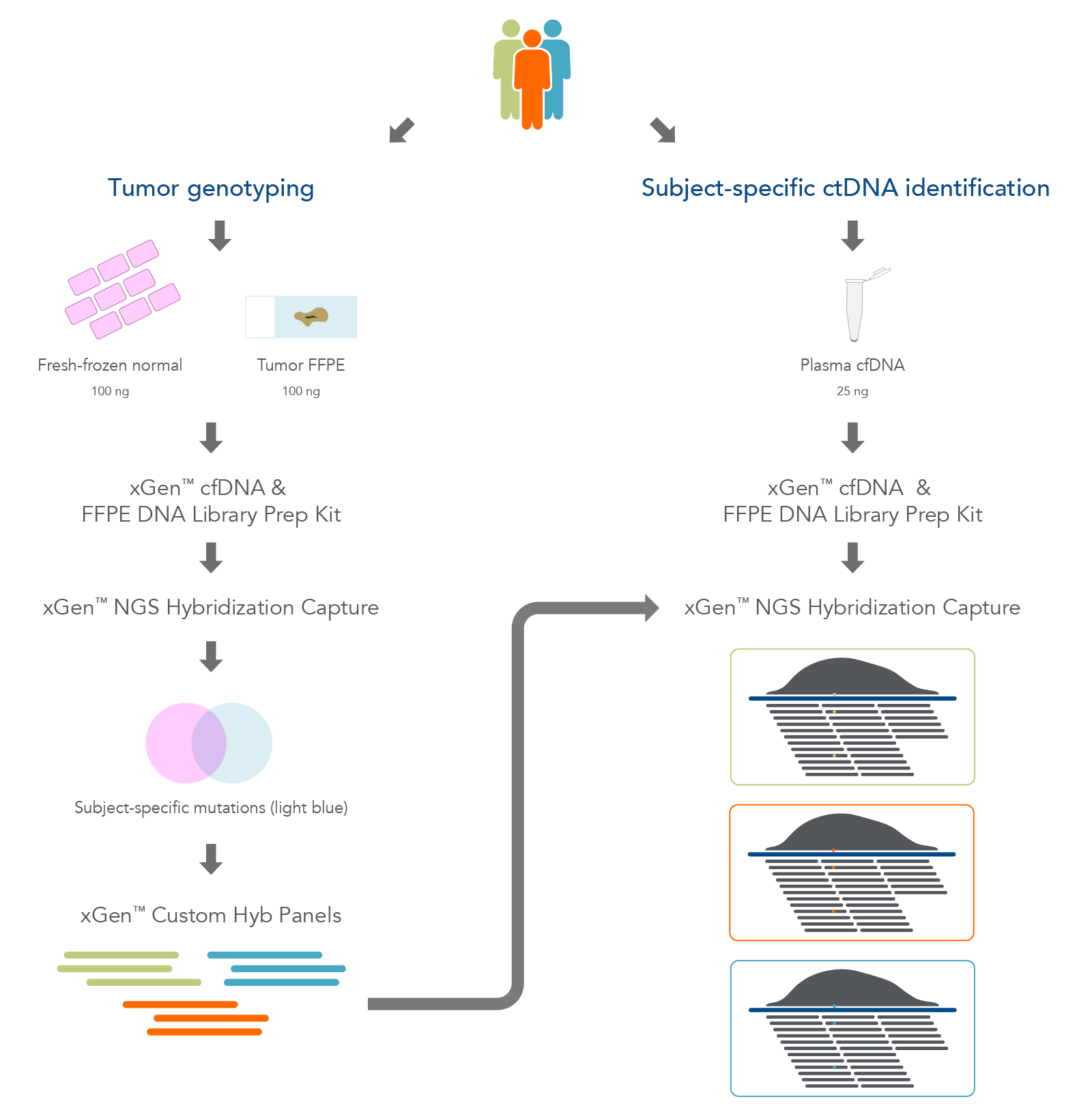
Figure 1. Overview of the research workflow. Fresh-frozen normal tissue, tumor-derived FFPE tissue, and plasma cfDNA were extracted from three biobank samples. The fresh-frozen normal and FFPE tissues were used for hybridization capture to identify tumor-associated variants. Sequencing was used to design subject-specific xGen Custom Hyb panels for use in targeted deep sequencing of the plasma cfDNA.
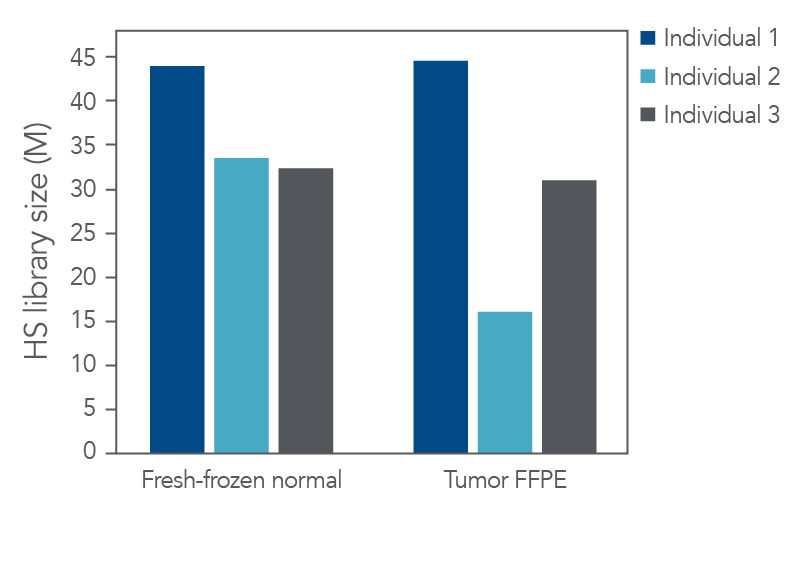
Figure 2. High-quality sequencing libraries from tumor and normal samples. Libraries derived from fresh-frozen normal and FFPE-tumor biobank samples (n = 1) were generated with the xGen cfDNA & FFPE DNA Library Prep Kit from 100 ng of input material. Libraries were captured in single-plex with a custom 2.2 Mb xGen Pan-Cancer Hybridization Panel. Libraries were pooled and sequenced on a NextSeq™ 500 (Illumina) instrument. Reads were subsampled to 140 M reads per library and mapped using BWA (0.7.15) [2]. Libraries were then deduplicated based on start-stop position using Picard (2.18.9) or fgbio (0.7.0) [3] as described in the xGen cfDNA & FFPE DNA Library Prep Kit Analysis Guidelines. HS library size was calculated using Picard [4]. Libraries and captures from both figures were amplified using KAPA 2x HiFi PCR Mix, as these data were generated prior to the release of xGen 2X HiFi PCR Mix.
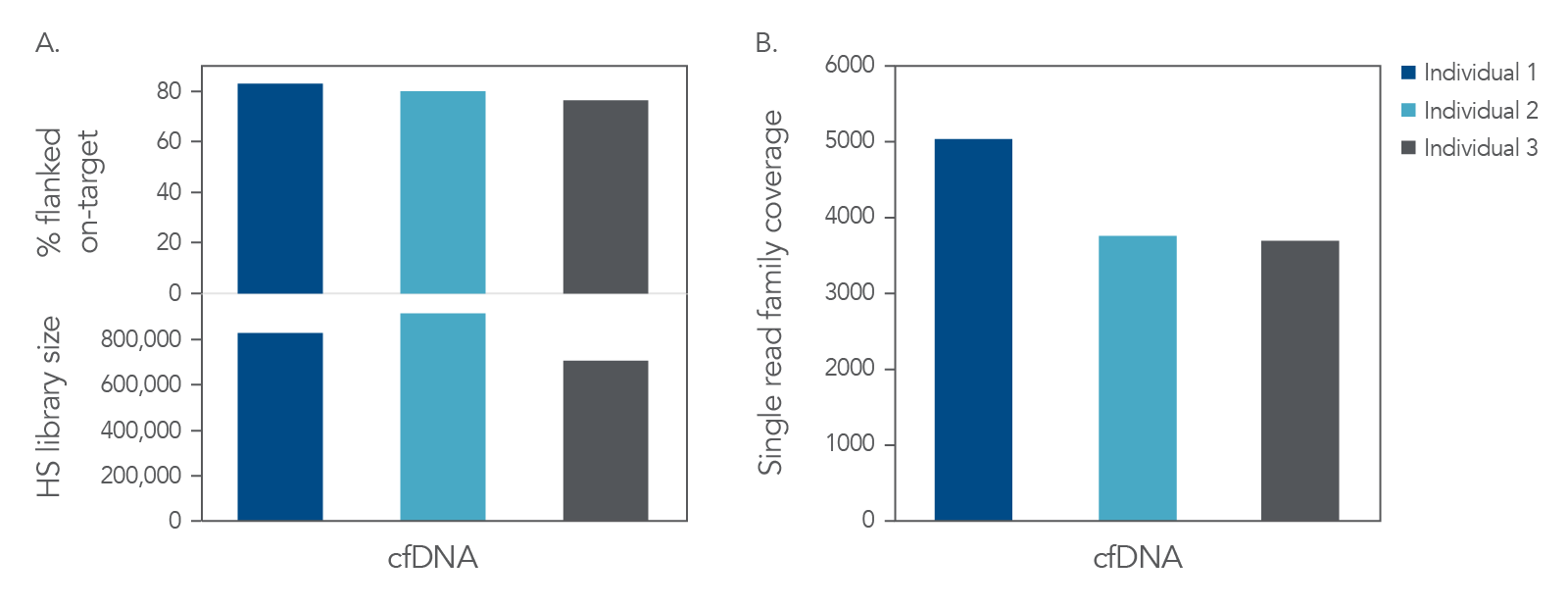
Figure 3. High complexity and coverage sequencing data from cfDNA. Libraries were generated with the xGen cfDNA & FFPE DNA Library Prep Kit from 25 ng of cfDNA material (n = 1). Libraries were captured in single-plex with custom subject-specific xGen Custom Hyb Panels. Libraries were pooled and sequenced on a NextSeq™ 500 (Illumina) instrument. Reads were subsampled to 40 M reads per library and mapped using BWA (0.7.15) [2]. Libraries were then deduplicated based on start-stop position using Picard (2.18.9) or error-corrected with single read families using fgbio (0.7.0) [3] as described in the xGen cfDNA & FFPE DNA Library Prep Kit Analysis Guidelines. (A) On-target rate was calculated using the standard on-target rate metric (PCT_SELECTED_BASES). HS library size and (B) coverage were also calculated using Picard [4]. Libraries and captures from both figures were amplified using KAPA 2x HiFi PCR Mix, as these data were generated prior to release of xGen 2x HiFi PCR Mix.
Ordering
Resources
References
- Beaudry MS, Wang J, Kieran TJ, et al. Improved microbial community characterization of 16s rRNA via metagenome hybridization capture enrichment. Front Microbiol. 2021;12:644662.
- Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754-1760.
- fgbio, GitHub repository. https://github.com/fulcrumgenomics/fgbio
- "Picard Toolkit." Broad Institute, GitHub repository: Broad Institute; 2019. https://github.com/broadinstitute/picard